Introduction of PIANO
PIANO is a prediction framework dedicated for transcriptome-wide Ψ RNA methylation site prediction using a machine learning approach. Besides the conventional sequence features that used on all three previously published Ψ relevant predictor, a high-accuracy Ψ site classifier, PIANO was established by integrating 42 additional genomic features. When tested on six independent datasets with four different Ψ profiling technologies (Ψ-seq, RBS-seq, Pseudo-seq and CeU-seq) as benchmarks, PIANO achieved an average AUC of 0.955 and 0.838 under the full transcript or mature messenger RNA models, respectively. Compared to the previously published models PPUS (0.713 and 0.707), iRNA-PseU (0.713 and 0.712) and PseUI (0.634 and 0.652), PIANO adds a marked improvement to the accuracy of existing Ψ sites prediction. Moreover, to help the users to explore underlying regulatory mechanisms of computationally predicted Ψ sites, PIANO firstly links the putative Ψ sites with various post-transcriptional regulation analysis by systematically annotates Ψ sites with miRNA-targets, RBP-binding regions, and splicing sites. Finally, a concise web server was built for academic users with simple and clear instruction, and 4303 experiment-validated human Ψ sites were also collected in the web server.
PIANO takes two kinds of format as input file.
(1): The tab-delimited txt format, which contains genomic position information (genome assembly: hg19) with four columns: chromosome, start position, end position, and strand.
Example:- 20 3204013 3204013 +
- 1 6710854 6710854 +
- 3 113804521 113804521 +
- 19 54819023 54819023 -
(2): The standard FASTA format.
Example:- >test1
AUGGGGGUGGAACUCAUGAUGGAAUUGGAGCCUUUACAAGGGAAUGAAGA GACAAGAGCUCUCUUUAUGCCACGUGAGGAUACAGCAAGGCCCCAAUCUG CAAGCCAGGAAGAGUCGUCACGAGAACCAGACCAUGCAGGAACUCUGAUC
GUGGA - >test2
UACUAAUUUUCAAAGGCGGGGUUCUGCCAGGCAUAGUCUUUUUUUCUGGC GGCCCUUGUGUAAACCUGUCUUUCAGACCUUGUUGGACAUCCCGUACAAU CAAGAUGUUCCUGUAUGUUGUUUGCAGUCUGGCGGUUUGCUUUCGAGGAC
UAUU
(3): Users can either use the example file we provided or upload their own file.
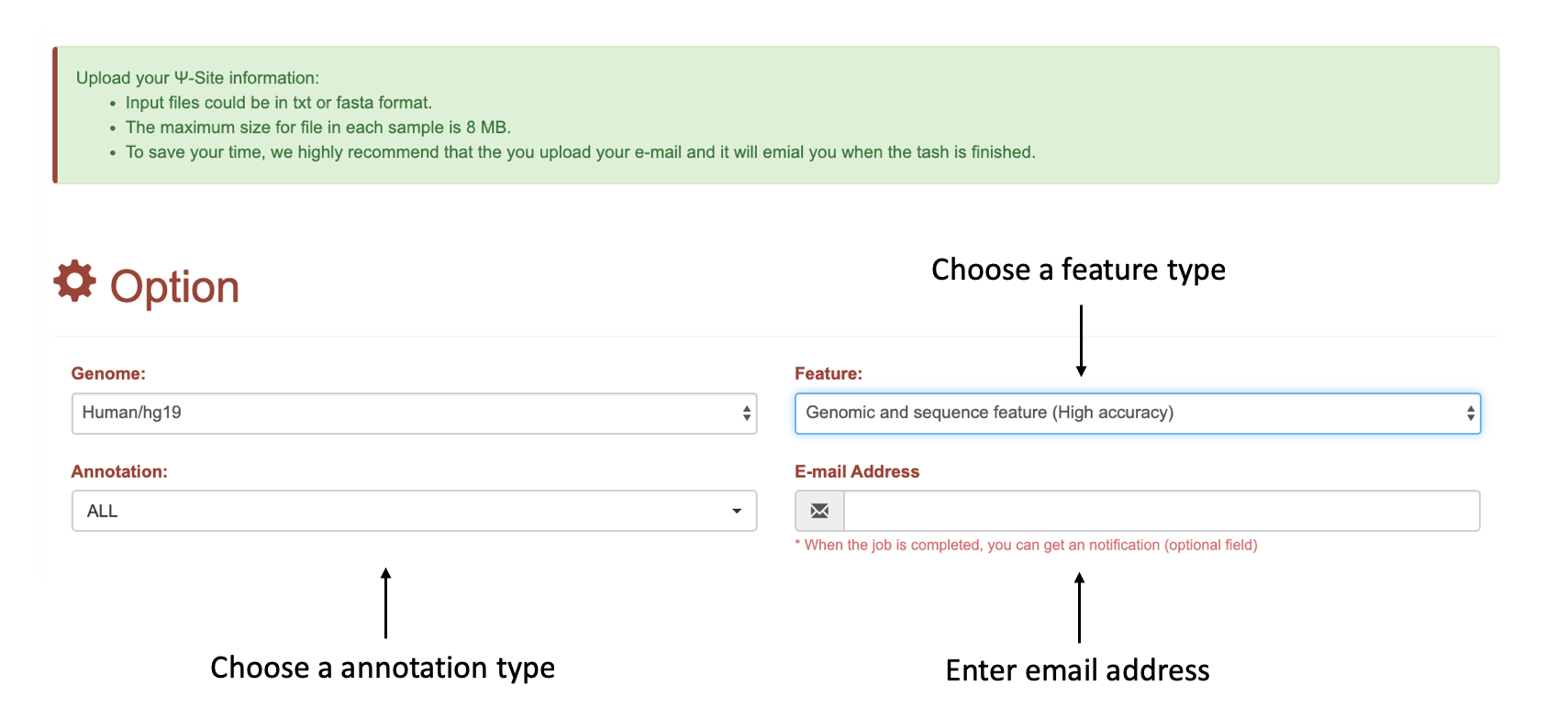
(1): Chose the feature and annotation types to start the analysis.
* The email address is strongly recommended, as the analysis may take some times and users can get notification email when their job is finished.
When users submit their job, an unique job ID will be created. Users can wait in this page for their job to be finished, or use the job ID to retrieve their jobs.
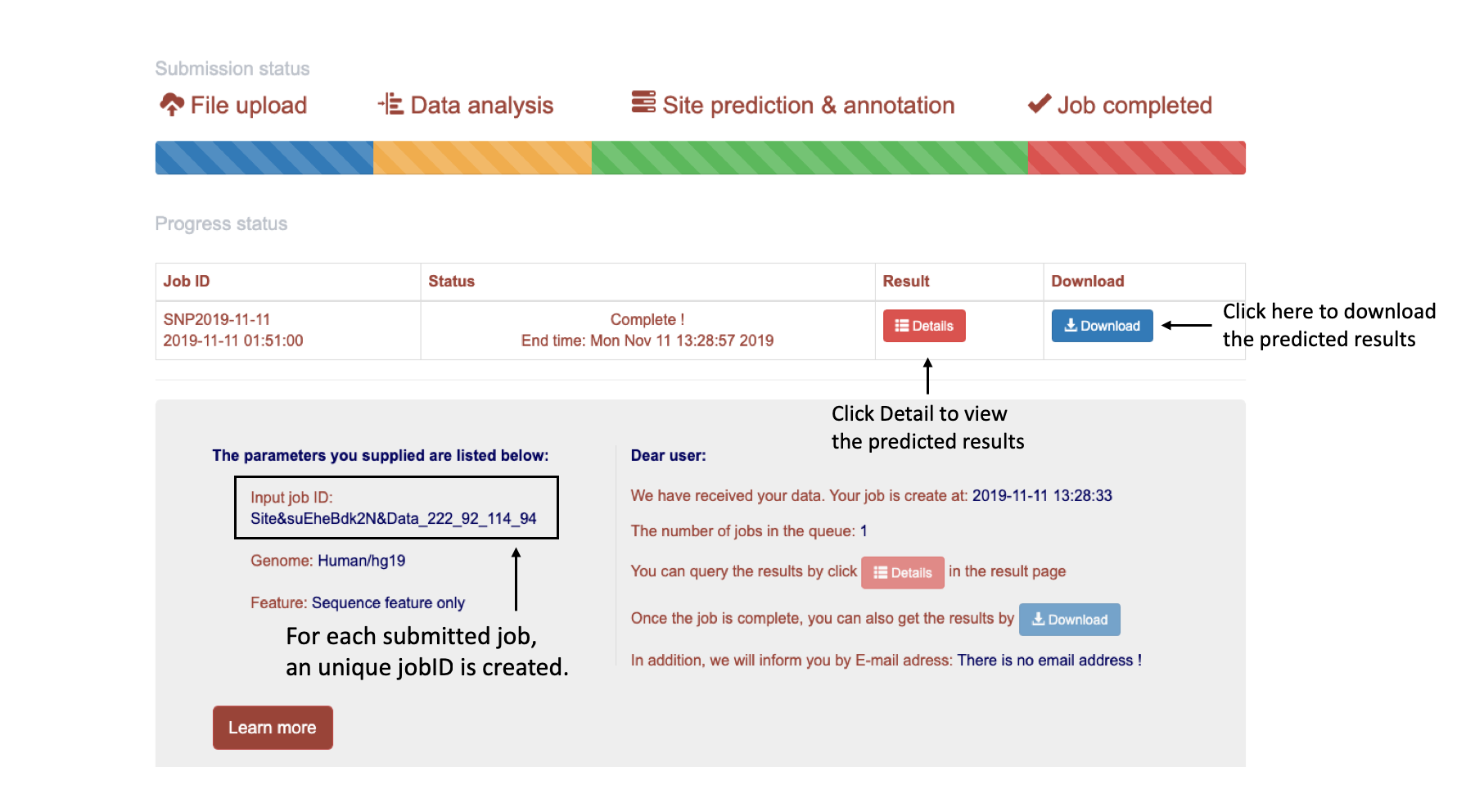
When users leave the job page or lose the page on accident, the job ID can be used to retrieve their job.
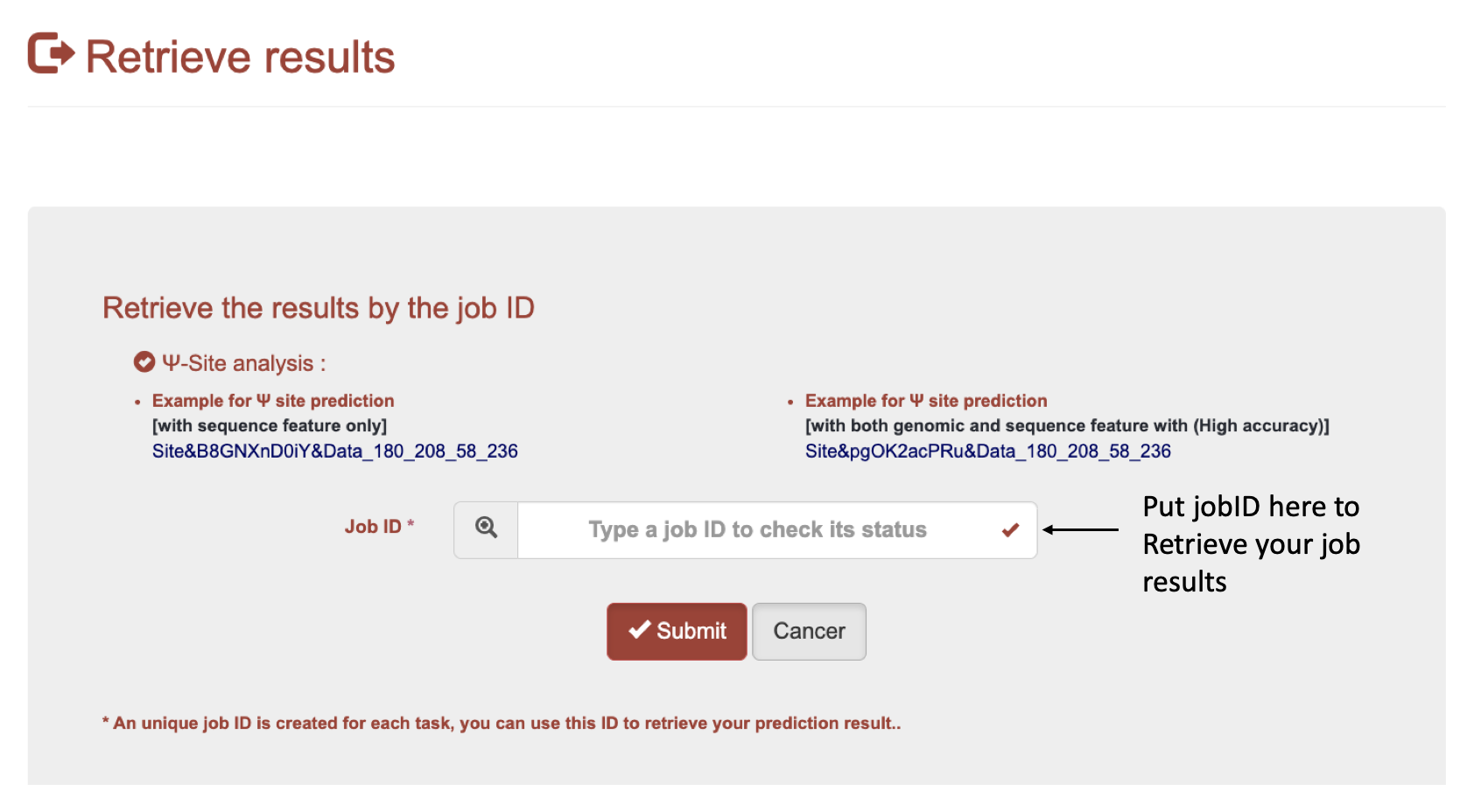
For users upload genomic position information using both genomic and sequence-derived features.
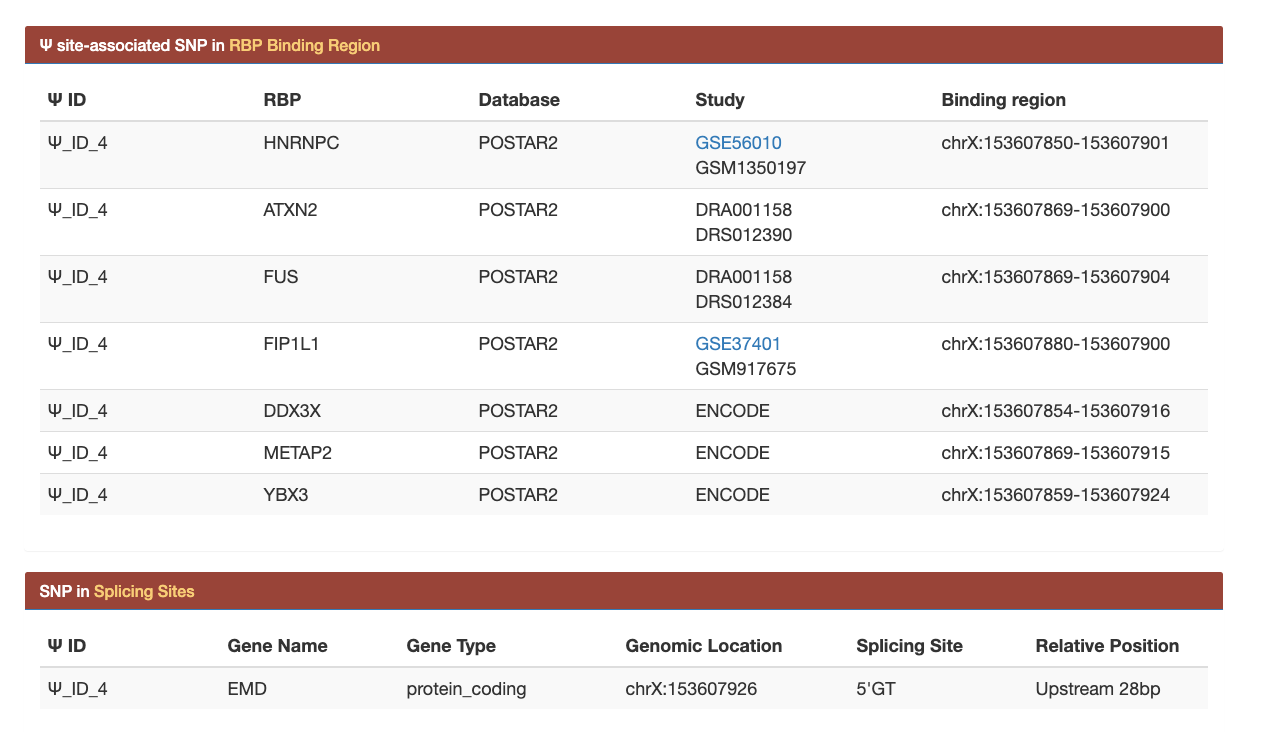
(1): For users upload genomic position information using both genomic and sequence-derived features, an overall summary table is provided. Users can view the basic information of putative Ψ sites with various annotations, such as gene symbol, likelihood ratio (LR), confidence level, and the number of related post-transcriptional analysis.
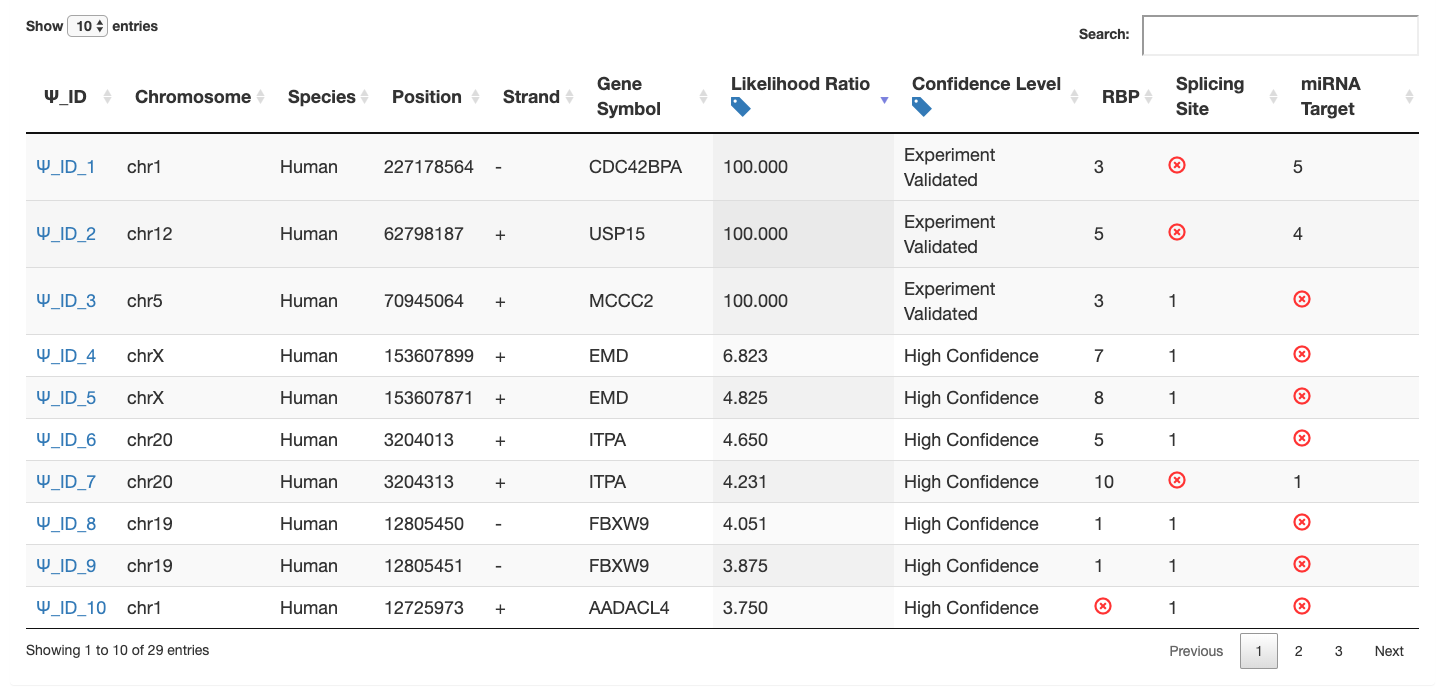
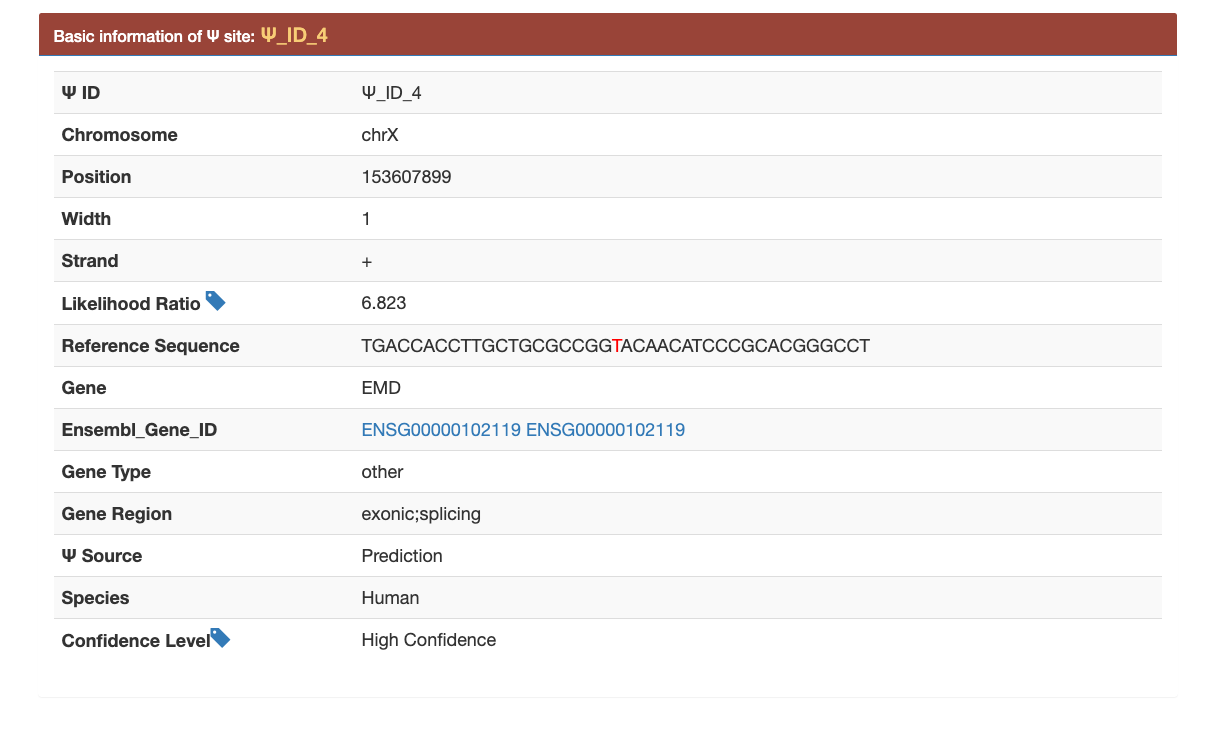
(2): By clicking the Ψ_ID, users can view the detail information of each putative Ψ site.
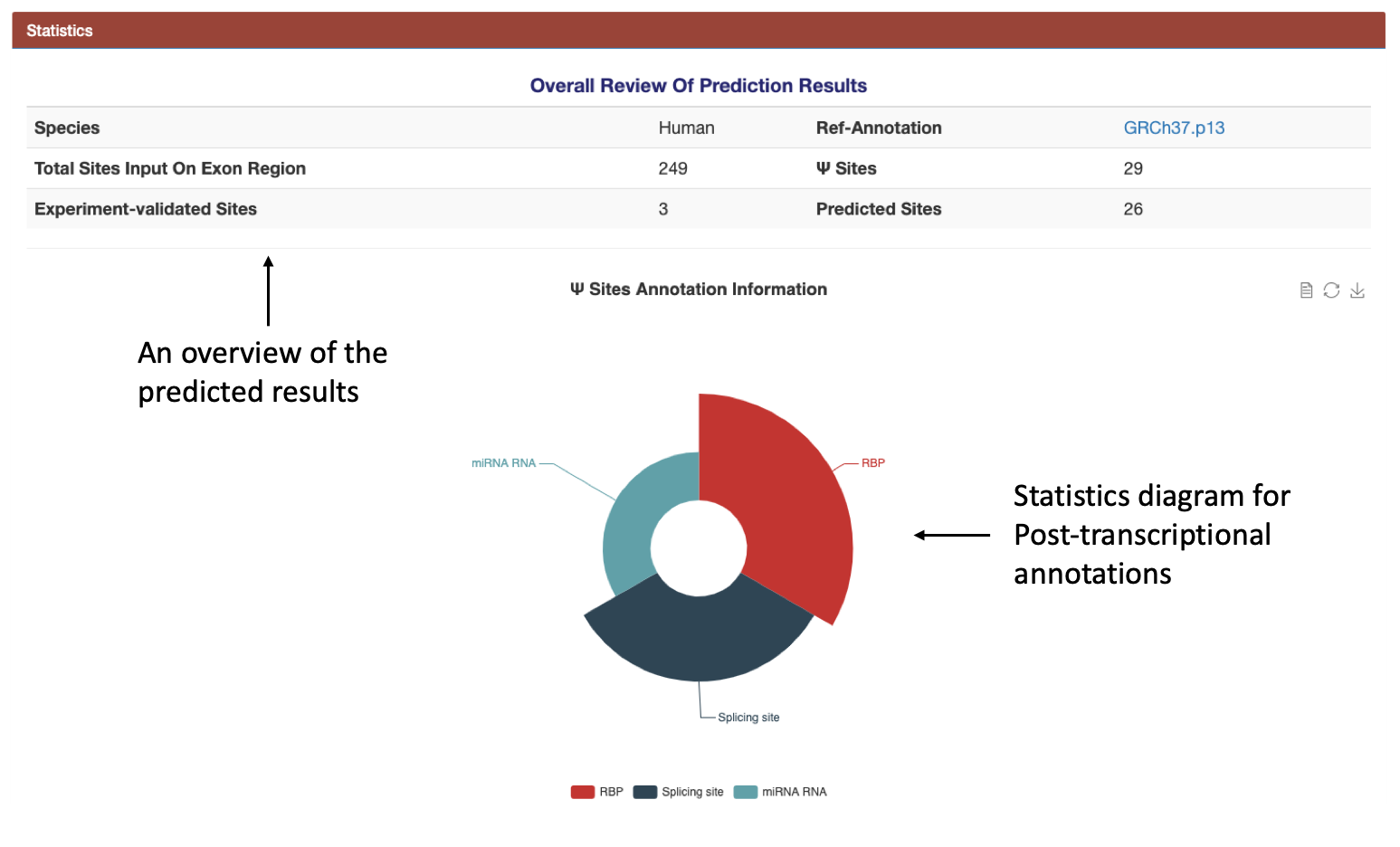
(3): The statistic table and diagram are provided for users to view their results clearly.
For users upload sequence information using sequence-derived features.
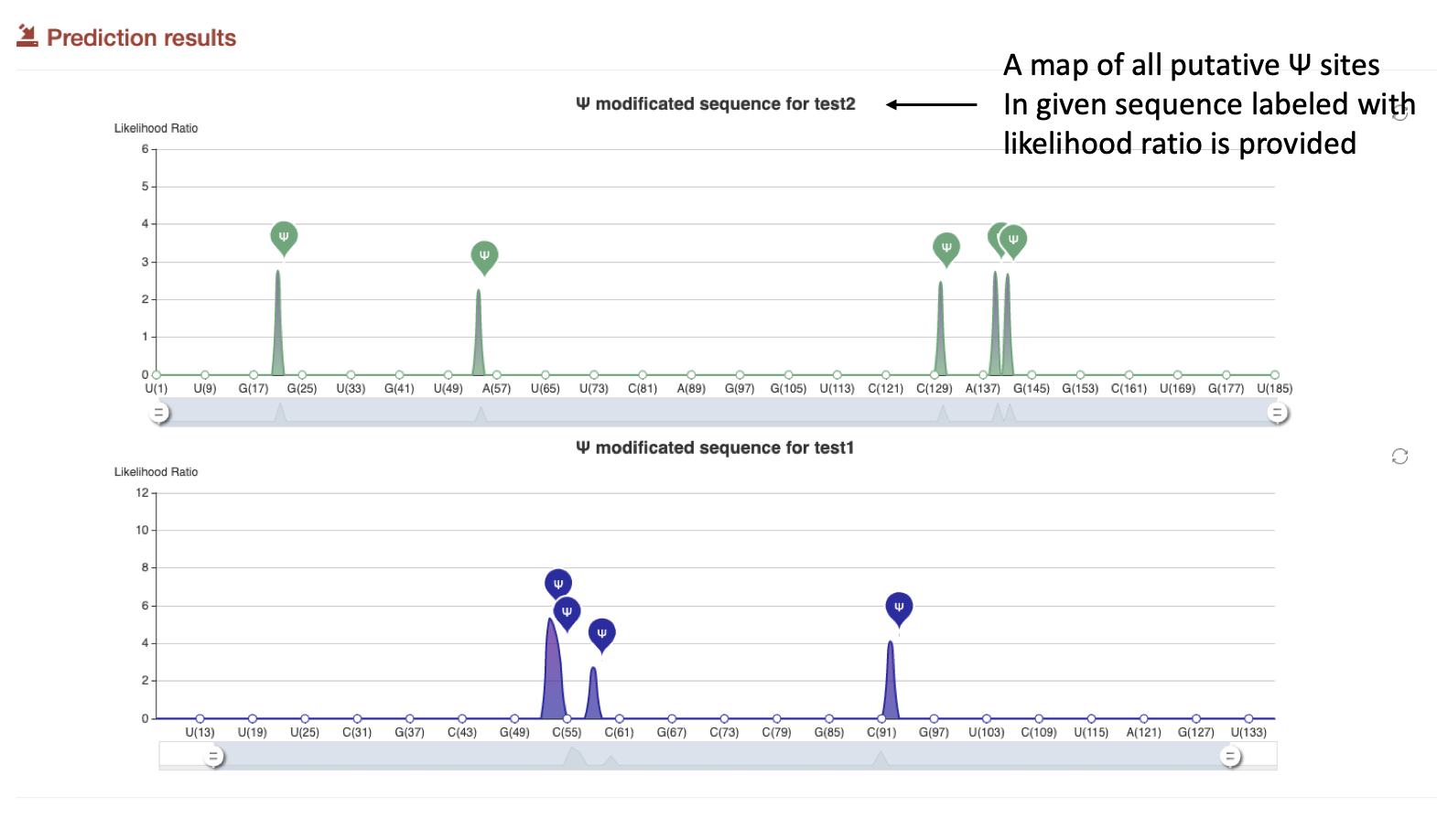
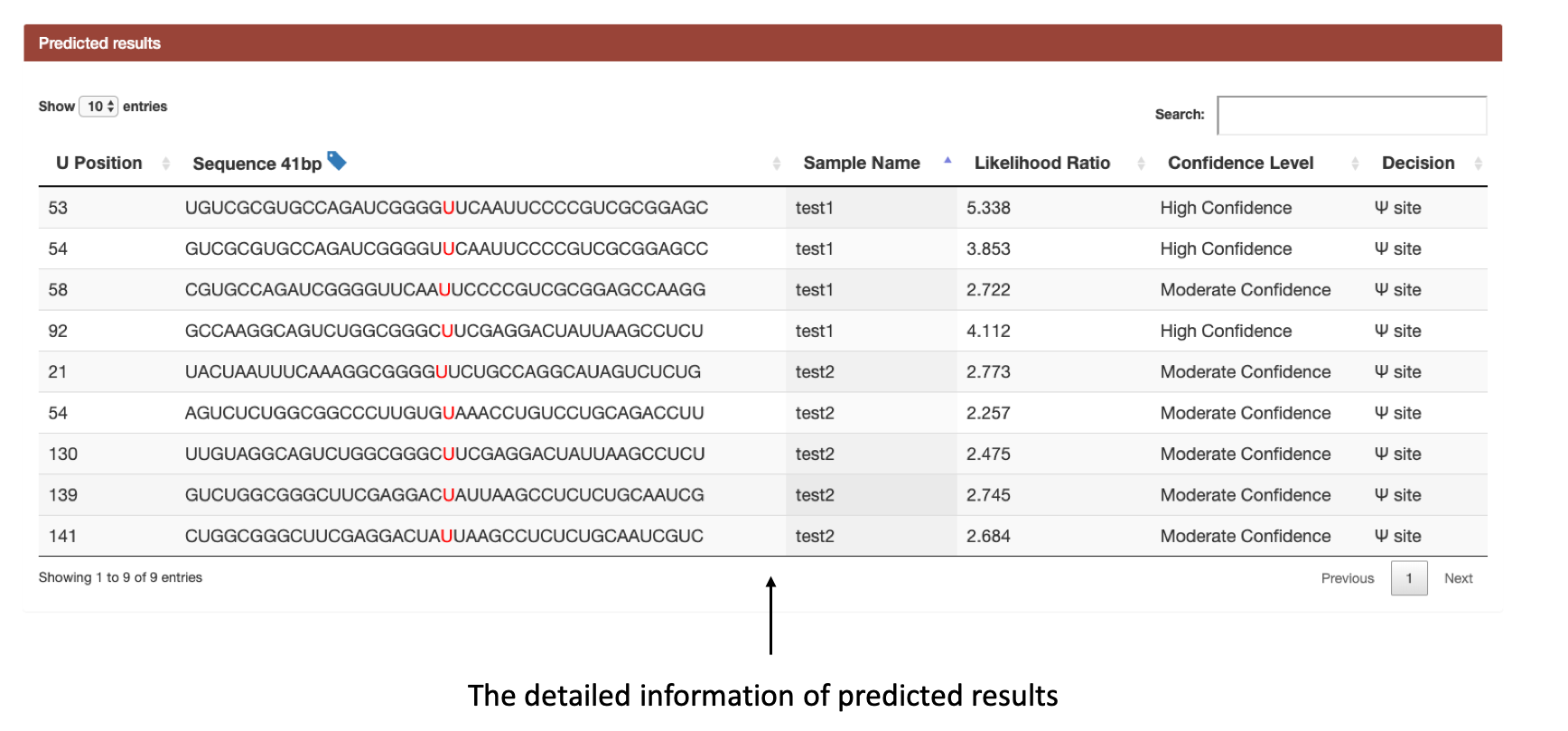
(1): Firstly, a map of all putative Ψ sites in given sequence is provided, users can know the position of each Ψ modification clearly. The detail information of each computational predicted Ψ site are then listed, with 41 bp sequence, sample name, likelihood ratio, and confidence level.